
Kind of a big deal: 10 golden-age papers that changed human genetics
Looking back at more than two decades of human genomics


In my conversation with Erich Schwarz we discussed how large model organisms loomed in the 20th century, from the essential role of Drosophila melanogaster in classical Mendelism to mice lines in medical genetics. But the draft human genome’s publication in 2000 also heralded a renaissance in the study of our own species, as cutting-edge genomic tools and methods often debuted and were first tested on human datasets. This century’s first few decades have been the golden age of human genetics, with more data at our fingertips than we could even have imagined as late as 1995, when it was still possible to find geneticists debating whether the human genome could in fact be sequenced.
What follows are ten papers that came out in the century’s first two fervent decades. All have materially shaped my understanding and practice of human genetics. This is in no way a comprehensive list, and readers who have followed me over the decades will see how these particular publications reflect my own idiosyncratic fascinations. Nevertheless, I would maintain that enough of them are truly seminal to form a core reading list reflecting what we’ve learned going into the genomic era's third decade.
And although I recognize papers in the field lie beyond the reading tastes of many of my loyal subscribers, I still think a passing awareness of each of these recent leaps forward in our understanding is warranted for anyone living through this golden age. If popular book-length treatments of these new insights and discoveries existed, I would be the first to recommend them. And since they do not yet, here are the papers themselves and my promise that I’ll keep chipping away at covering their contents for the lay reader, Substack piece by Substack piece. For the truly committed and the specialists, I’ve included a subsequent companion paper on each topic. Feel free to share your own hit list in the comments.
(2002) Detecting recent positive selection in the human genome from haplotype structure. Pardis Sabeti’s 2002 paper kicked off the age of tapping genomic-scale data to answer old evolutionary questions like: what regions of the genetic map show signatures of natural selection? Sabeti and her coauthors’ innovation was to create an inferential workhorse by surveying genetic variants clustered together within the genome. These variants together define haplotypes, unique segments in the genome. They looked for haplotypes that were both common, indicating positive selection pushing their frequency upward, and very long, a tell that the rapidity of the sweep had outrun recombination. The method takes advantage of the dynamic of new evolutionarily favored mutations spreading so fast that they also drag adjacent and unrelated markers along upward in frequency. This synchronicity yields a long tract of correlated variants, the haplotype, whose length reflects the strength of selection. Though the method draws on elegant and simple classical genetic principles, only with the tsunami of data generated by 21st-century technology was there enough power to drive the engines of these analyses (since 2002 the field has covered a great distance, including exploring complex traits and selection along the whole genome like 2020’s Polygenic adaptation: a unifying framework to understand positive selection).
(2005) SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. This 2005 paper combines statistical genetics, animal models and molecular biology to find the biggest locus that impacts variation in human pigmentation. It was published after Armand Leroi’s Mutants: On Genetic Variety and the Human Body, where he observed in the final chapter that even as late as the first decade of the 21st-century, geneticists lacked a good grasp on the genetic architecture of something as simple as skin color. This paper, published just a few years later, was the first result in a batch of findings between 2005 and 2010 that mapped most of the major genetic loci responsible for variation in pigmentation. These results allowed researchers to unlock the power of forensic genomics in the 2010’s, enabling paleogeneticists to reconstruct the physical appearance of humans who died thousands of years ago. The media often terms the gene identified in this paper the “gene that makes you white,” but I carry two copies of the “white” variant and can attest that I am quite brown, so it’s a bit more complicated than that. (With the identification of markers implicated in skin color variation, later researchers began to explore their adaptive history as in 2020’s The evolution of skin pigmentation-associated variation in West Eurasia).
(2006) A Map of Recent Positive Selection in the Human Genome. Applying and extending the methods in the 2002 Sabeti paper, this publication showed how statistical genetic techniques can leverage hundreds of thousands of markers across hundreds of individuals to explore signatures of evolutionary history. Earlier work studying selection in human genes tended to target and focus on specific regions that might be interesting, but A Map of Recent Positive Selection in the Human Genome lit the way for labs to begin churning out promising candidates for further exploration, ushering in the age of industrial-scale genomic analysis. (The 2006 paper opened up the path for others, but follow-ups like 2014’s Genome-wide signals of positive selection in human evolution illustrate the many subtleties of constructing one-size-fits-all statistics to assess selection at the genomic level).

Iberian Neanderthal man (2010) A Draft Sequence of the Neandertal Genome. I think it is defensible to say that this paper heralded the arrival of the ancient DNA age and hammered home the relevance of modern paleogenetics for understanding human evolution (the last author Svante Paabo went on to win the Nobel Prize). Though published after the sequence of an ancient Greelander, the fact that this work revealed the genome of a Neanderthal and proved their contribution to the ancestry of most humans alive today was transformative. Instead of a simple out-of-Africa story some 100 thousand years ago, the origin of our species became a complicated network of relationships going back nearly a million years. In the decade before this publication, it was possible to wonder if Neanderthals could even speak; after it, no one doubted the capabilities of our cousin lineage, which had become part of our heritage. (Today the admixture of Neanderthals in our lineage is taken for granted, so more of the focus is on what Neanderthal ancestry does in our genome, as in 2022’s The contribution of Neanderthal introgression to modern human traits).

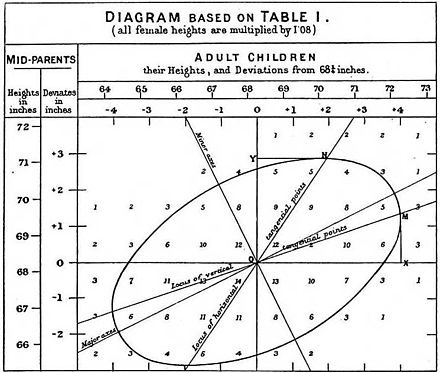
(2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Height has long been understood to be heritable; the variation in the population of this trait is due mostly to variation in the genes carried across the population. Height was one of the classic illustrations of Francis Galton’s concept of regression, with the height of sons on the y-axis correlating with the height of the fathers on the x-axis. This 2010 paper began to nail down the concrete molecular basis of height as a heritable trait, using genome-wide association to track more than 600 variants that differed between tall and short individuals, and showing that not only did they correlate with height but that these markers also associated with known skeletal genes. Today many more markers are known, but the overall map of how to illuminate the architecture of a quantitative genomic trait dates to this paper. (The field of height genetics has advanced far enough now that scientists can predict the trait from genes alone, as in 2020’s Polygenic Scores for Height in Admixed Populations).
(2014) Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. In 2014, researchers discovered that Denisovans, a population known only from ancient DNA, contributed one of humanity’s major altitude adaptations. Theoretically, this should be expected; when you have a long-resident population absorbed by newcomers, bespoke local adaptations should be favored in the admixture. A decade on, we’ve observed many other instances of this phenomenon, and it is clear that most of Tibetan ancestry arrived in high latitudes only within the last 5,000 years, so with hindsight, the selection for Denisovan EPAS1 shouldn’t be that surprising, but at the time it was an amazing discovery. (With EPAS1 discovered, now geneticists are searching for many more variants in populations with a great deal of archaic admixture, like Melanesians in this 2024 preprint, Denisovan admixture facilitated environmental adaptation in Papua New Guinean populations. In 2019 I also recorded a podcast with the author of the EPAS1 paper).

Mesolithic Western European forager (2014) Ancient human genomes suggest three ancestral populations for present-day Europeans. Combining ancient DNA with large-scale modern genomic datasets, this paper sketched out the general structure and origin of European populations as we know them today. Though later work clarified details, the general outline has yet to require rewriting. Europeans descend from Mesolithic hunter-gatherers, from eastern Mediterranean farmers, and from a population with Siberian affinities. A year later, DNA results from a burial pit in Ukraine would confirm that the Siberian signature came from Indo-Europeans, filling in the tableau’s last major detail. (That final piece of the puzzle came into focus with 2015’s Massive migration from the steppe was a source for Indo-European languages in Europe).
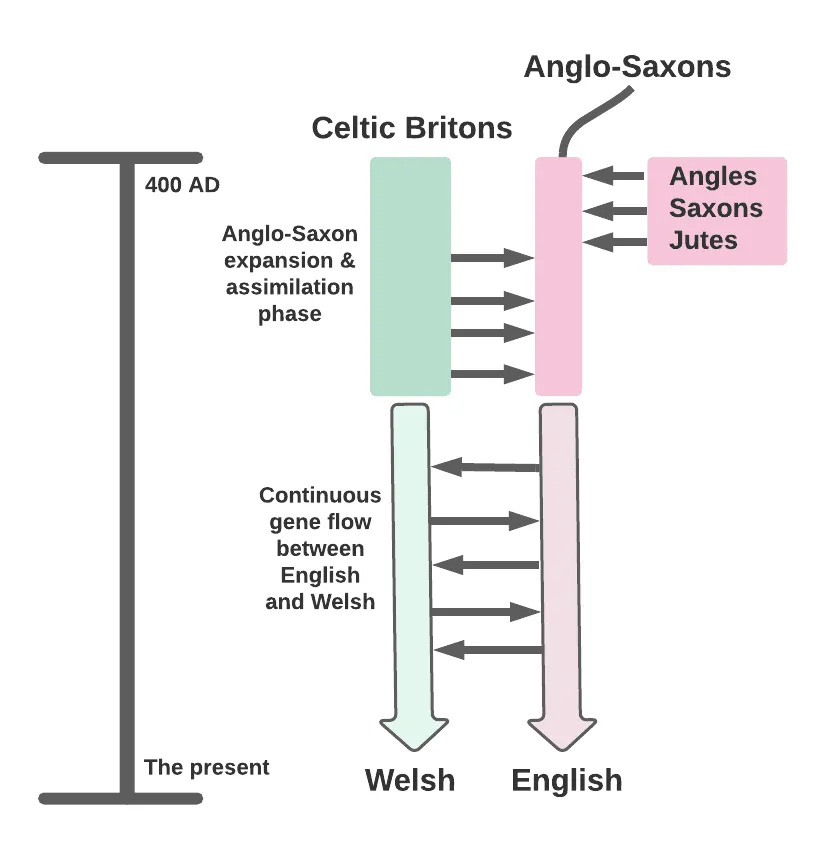
(2015) The fine-scale genetic structure of the British population. This 2015 paper was the culmination of data collection and collaborative work by many researchers. It reconstructed British population history and surveyed British population structure using more than 2,000 individuals whose genomes were typed and whose documented provenance was minutely traced back to their four grandparents’ ancestral home counties. The dense genotype data allowed for the utilization of very advanced haplotype-based methods of inferring ancestry and relatedness, which answered questions of deep interest like the proportion of Anglo-Saxon ancestry across the island and the relatedness of British people to various European populations. (2022’s The Anglo-Saxon migration and the formation of the early English gene pool built on the foundations of the 2015 paper, adding ancient DNA and whole-genome analysis to the picture. See also my podcast with the driving force behind the 2015 paper and the project generally, Walter Bodmer).
(2015) A global reference for human genetic variation. Most have heard of the 1000 Genomes Project, and the 2015 paper published by the consortium remains an essential reference for grasping the nitty-gritty of variation in the human genome. If you need to look up the number of polymorphisms in a given population to get a read on whether your sequencing effort makes sense, this is the paper you would consult and cite in any reference. Some scientific research targets very narrow and precise hypotheses, but A global reference for human genetic variation genuinely stands as more of a general resource for other researchers (2020’s Insights into human genetic variation and population history from 929 diverse genomes applies the same methods of the 1000 Genomes Project to the decades-old Human Genome Diversity Project dataset; its results broadly similar, but materially different specifically due to the less cosmopolitan population set selected by L. L. Cavalli-Sforza and his collaborators in the 20th century).
(2016) Genome-wide association study identifies 74 loci associated with educational attainment. Many follow-ups to this paper have identified further genetic markers associated with educational attainment, but this was the publication that broke open the door to this controversial field of study. Though behavioral, cognitive and social traits have long been understood to be heritable, their genetic architecture, with effects distributed across thousands of genes, remained difficult to detect with standard genetic methods in the 20th century. Genomics and modern computation eased the development of new methods and the aggregation of enough data to pick up weak but definite signals across thousands of markers. (A 2022 paper, Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals, increases the number of markers to nearly 4,000, with a predictive power of about 15% of the variation in educational attainment. And here’s my podcast with two of the authors).


There's been similar research to the Anglo-Saxon one in Poland.
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03013-9
Polish archaeogenetics is a bit complicated by the common practice of cremation, but we now have definitive proof that in the first centuries AD the area around the Vistula was settled by a people from Scandinavia, that was different from medieval and current populations of Poland.
This confirms at least some of the Gothic peoples' own migration story, as reported by Jordanes, and should make a few people who have theorised that European 'Barbarians' were a formless blob (a 'Barbaricum') until brought into contact with Romans embarassed.
There are still a lot of unanswered questions (who or what the Przeworsk culture people are and how they're related to Goths, Vandals etc.) but it's a big indicator that ancient 'barbarian' sources can't just be discarded, and that there's quite a bit more continuity in tribal identity than some people have theorised.
Love this list. Got lots of reading to catch up on…